

GONADOTROPIN RELEASING HORMONE (GnRH) – THE PRINCIPAL REGULATOR OF REPRODUCTION
The Discovery of GnRH
GnRH was isolated from porcine hypothalami and structurally identified as a decapeptide (pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly·NH2) five decades ago (4-6). This decapeptide was shown to potently stimulate LH and FSH release from the pituitary in a number of mammalian species (6,7). Early literature referred to this peptide as the ‘Luteinizing Hormone-Releasing Hormone (LH-RH)’, but more recently, it is widely referenced as Gonadotropin Releasing Hormone (GnRH) -reflecting the stimulatory role on the secretion of both gonadotropins – i.e., LH and FSH (8).
Diverse forms of GnRH and its receptor exist among vertebrates, with over twenty primary structures across species, suggesting that GnRH system developed early in the evolutionary sequence (9,10). The GnRH structure was first identified in mammals and is therefore referred to as GnRH I (9,11). Subsequently, another structurally different vertebrate GnRH sequence was first identified from chicken brain -this is now referred to as GnRH II (pGlu-His-Trp-Ser-His-Gly-Trp-Tyr-Pro-Gly-NH2) (10,12). A third form has also been described in fish – GnRH III (9,12). In mammals, hypophysiotropic functions are limited to GnRH I, therefore in the human context GnRH I is referred to as GnRH (13) and we will use this terminology for this review.
Neuroanatomy of GnRH Neurons
GnRH neurons originate in the medial olfactory placode during embryological development and migrate along the olfactory bulb to their final positions within the hypothalamus (14,15). A number of factors contributing to this GnRH neuron migratory process have been identified: anosmin-1 (the product of KAL gene) (16), neuropilins (17), leukemia inhibitory factor (18), fibroblast growth factor receptor 1 (19), fibroblast growth factor receptor 8 (20), polysialic form of neural adhesion molecule (PSA-NCAM) (21), among others (22). Defective GnRH migration leads to Kallmann syndrome, characterized by hypogonadotropic hypogonadism due to GnRH deficiency and anosmia (15). Mutations in prokineticin genes (PROK1 and PROK2) lead to hypogonadotropic hypogonadism without anosmia, suggesting that factors other than suboptimal migration can also lead to functional deficiencies in GnRH (15,23,24).
GnRH cell bodies are located in the medial preoptic area (POA) and in the arcuate/infundibular nucleus of the hypothalamus, forming a neuronal network with projections to the median eminence (25). GnRH is secreted from the median eminence into the fenestrated capillaries of portal circulation, carried to the anterior pituitary (25). In humans, the number of GnRH neurons has been estimated to range between 1000 to 1500 (9,14). The co-location of GnRH neurons with other central regulators allows the GnRH network to be influenced by a range of neuroendocrine and metabolic inputs (26).
GnRH Secretion and Pulsatility
Two distinct modes of GnRH secretion have been described: pulsatile and surge modes (26). Pulsatile mode refers to episodic release of GnRH, with distinct pulses of GnRH secretion into the portal circulation with undetectable GnRH concentrations between pulses. The surge mode of GnRH secretion occurs in females, during the pre-ovulatory phase, in which the presence of GnRH in the portal circulation appears to be persistent (26,27).
Direct pulsatile GnRH release was initially demonstrated in ovariectomized rhesus monkeys using serial samples of portal blood (28). Pulsatile pattern of GnRH secretion was demonstrated subsequently in humans through serial blood sampling during pituitary surgery (29). Abolishment of LH pulses by GnRH antisera (30,31) and its reestablishment with GnRH analogues (30) suggest that LH pulses are determined by the underlying GnRH pulsatility. The LH pulsatility was first detected during an attempt to validate a radioimmunoassay to measure serum LH in rhesus monkeys, where marked variations in LH levels was noticed (32). Further studies confirmed the pulsatile nature of LH secretion (33-35). In women, the frequency and amplitude of LH pulses were noted to be dependent on the menstrual cycle phase, with pulses every 1 to 2 hours during the early follicular phase eventually merging into a continuous mid-cycle surge, and decreased pulse frequency to every 4 hours during the luteal phase (36). In humans, LH pulse frequency is used as a surrogate of GnRH pulsatility, as ethical considerations and technical challenges preclude sampling of hypophyseal blood or cerebrospinal fluid to measure GnRH concentrations directly (37,38).
The importance of GnRH pulsatility on LH and FSH secretion was first demonstrated in rhesus monkeys, where endogenous GnRH secretion was abolished by hypothalamic radiofrequency. Pulsatile GnRH reinstated gonadotropin secretion in these animals, whereas continuous GnRH only elicited a transient response. Moreover, the switch from continuous to pulsatile GnRH administration allowed recovery of gonadotropin secretion (39).
GnRH neurons coordinate their activity, but the precise mechanism of this remains unclear (27,40), and is the subject of continuing investigations. Episodic multi-unit electrical activity at medial basal hypothalami (MBH) is correlated with LH release, suggesting that ‘GnRH pulse generator’ is anatomically located at MBH – or closely linked to it neurohormonally (41,42). GnRH neurons show intrinsic electrical pulsatility. GnRH cell lines derived from mouse hypothalamic and fetal olfactory placode GnRH neurons both demonstrate intrinsic pulsatility in vitro (26,43,44). Functionally, the ‘GnRH pulse generator’ relies on complex relations between glutamatergic cells, GnRH and other neurons, and likely other elements are involved, of which the kisspeptin-neurokinin B-opioid pathway may have a pivotal intermediary role in the regulation of GnRH pulsatility (45).
Differential Regulation of LH and FSH
The stimulatory effects of GnRH on LH and FSH secretion are not identical (46). FSH secretion is more irregular than LH in both humans and sheep, which is essentially related to the pulsatility and different stimulatory effects of GnRH, but other factors also might be relevant, such as differences in LH and FSH storage (more scarce for the FSH), existence of different gonadotropes subpopulations, or diverse response times to GnRH (47). In ovariectomized sheep administered GnRH antisera, pulsatile secretion of LH was completely inhibited (undetectable LH levels within 24 hours), while the FSH concentration fell more slowly and remained detectable (30). It has been estimated that 93% of the GnRH pulses were associated with FSH pulses and, unlike LH, a constitutive secretion of FSH appears to exist (48). The frequency of GnRH input has been demonstrated to selectively regulate gonadotropin subunit gene transcription: rapid GnRH pulse rates increase α and LH-β and slow GnRH pulse frequency increases FSH-β gene transcription (49-51). Moreover, with progressive increases in GnRH frequency (from one pulse every 120 to 60 min, from 60 to 30 min, and from 30 to 15 min) in GnRH deficient men, mean LH rose concurrently with a decrease in LH pulse amplitude, while FSH remained unchanged (52).
Biological and Clinical Relevance of GnRH Pulsatility
Appropriate modulation of LH pulse frequency is essential for pubertal maturation and reproductive function. In infancy, LH pulsatile secretion is increased (often termed mini-puberty), likely reflecting pulsatile GnRH secretion, but soon becomes quiescent (53). This pre-pubertal suppression of HPG axis has been shown to occur in agonadal humans (54) and primates (55), suggesting that hypothalamo-hypophyseal factors play a role in post-natal quiescence of the reproductive axis, until puberty sets in.
The onset of pubertal maturation is heralded by the development of a pattern of steady acceleration in LH pulsatility (56). In children, higher basal and GnRH stimulated LH concentrations are observed in early childhood (<5 years). This is subdued mid-childhood (5-11 years) and increase thereafter with pubertal development (54,57). Conceptually, an abnormal reactivation of GnRH pulse frequency is the central mechanism associated with precocious or delayed puberty (14).
In women, the pattern of GnRH secretion is essential for the regulation of the menstrual cycle () (58,59). LH pulse frequency is slow in the luteal phase, and increasingly speeds up during the follicular and the pre-ovulatory phases, presumably reflecting changes in GnRH pulse frequency (60). Abnormalities in GnRH – and hence LH pulse frequency – are associated with a number of reproductive endocrine disorders. In hypothalamic amenorrhea, a condition associated with anovulatory amenorrhea and hypoestrogenism, LH pulse frequency (and by inference GnRH) is lower than expected for the prevailing steroid profile and is comparable to luteal phase pulsatility (37). LH pulse frequency in hyperprolactinemic women is also lower than in healthy women, requiring dopaminergic agonist preparations, such as bromocriptine to regulate prolactin secretion and restore LH pulse frequency (38). In polycystic ovary syndrome LH pulse frequency and amplitude are higher throughout the menstrual cycle in comparison to that observed in healthy women, contributing to chronic anovulation (61-64).

Figure 1.
Hormonal oscillations through the menstrual cycle. In the early follicular phase of the menstrual cycle, the initial increase in FSH stimulates follicular recruitment and maturation. The consequent secretion of estradiol (E2) selectively inhibits FSH release (needed for selection of the dominant follicle) and maintains rapid GnRH pulsatility during the late follicular phase. The persistent rapid GnRH pulses increase LH, which further stimulates E2 secretion, culminating in positive E2 feedback to produce the mid-cycle LH surge. During the LH surge, GnRH levels appear to be consistently elevated and remain elevated as LH declines, suggesting that the frequency of GnRH pulse has become very rapid or continuous, which results in desensitization of LH secretion (possibly the mechanism to terminate the LH surge). After ovulation, luteinization of the ruptured follicle results in progesterone secretion which reduces the frequency of GnRH pulses. With the demise of corpus luteum, E2, progesterone and inhibin levels fall, and the GnRH pulse frequency increases, leading to follicular maturation in the next cycle. (Adapted from: Marshall JC, Dalkin AC, Haisenleder DJ, Paul SJ, Ortolano GA, Kelch RP. Gonadotropin-releasing hormone pulses: regulators of gonadotropin synthesis and ovulatory cycles. Recent Prog Horm Res. 1991;47:155-187).
NEURONAL REGULATION OF GnRH SECRETION: THE KISSPEPTIN-NEUROKININ B-DYNORPHIN (KNDy) NEURONAL NETWORK
Whilst the central role attributed to GnRH remains undisputed, its effective function requires input from other neuronal networks. For instance, the absence of estrogen receptor alpha (ER-alpha) expression on GnRH neurons suggests the need for an intermediate signaling pathway to mediate gonadal steroid feedback (1). The discovery of kisspeptin signaling in neuroendocrine regulation of human reproduction revolutionized the current understanding of the HPG axis. Kisspeptin signaling pathway is increasingly recognized as essential for normal puberty, gonadotropin secretion, and regulation of reproduction (65-67). Other relevant kisspeptin roles have been identified such as regulation of sexual and social behavior, emotional brain processing, mood, audition, olfaction, metabolism, body composition, cardiac function, among others (68-74).
Discovery of KNDy Neuronal Network
KiSS1, the gene encoding kisspeptins, was first described in 1996 as a suppressor of metastasis in human malignant melanoma (75,76). This gene was discovered in Hershey and named in accordance with the famous chocolates ‘Hershey’s Kisses’; the inclusion of ‘SS’ is indicative of ‘suppressor sequence’. The KiSS1 gene maps to chromosome 1q32 and includes four exons of which the first two are not translated (77). The gene encodes the precursor 145 amino acid peptide, which is cleaved down to a 54 amino acid peptide. This peptide can be truncated to 14, 13 and 10 amino-acid peptides, all sharing the C-terminal sequence (78,79). These peptides are collectively referred to as kisspeptins – and Kp-10, Kp-13, Kp-14 and Kp-54 are suggested abbreviations for human kisspeptins (80). In 2001, kisspeptins were identified as ligands for the orphan G–protein receptor 54 (GPR54) (81-83), currently named KISS1R (80). KISS1R is localized to human chromosome 19p13.3 and it has five exons, encoding a 398-amino acid protein with seven trans-membrane domains (79,82). Upon binding by kisspeptin, KISS1R activates phospholipase C and recruits intracellular messengers, inositol triphosphate and diacylglycerol, which in turn lead to the release of calcium and activation of protein kinase C (82-84).
A reproductive role for kisspeptin in humans became apparent from patients with pubertal disorders which were associated with KISS1R mutations (85-87). A number of inactivating mutations of Kiss1 and Kiss1r have since been reported in animal models with phenotypes characterized by pubertal delay (88). An activating mutation in KISS1R has been described in a girl with precocious puberty: when compared to cells with wild-type transfected GPR54, cells with this mutation showed prolonged inositol phosphate accumulation and phosphorylation of extracellular signal–regulated kinase, suggesting extended activation of intracellular signaling by the mutant GPR54 (89). Missense mutations have also been reported in KISS1 gene in three unrelated children with central precocious puberty (90). Functional studies of these mutant peptides demonstrated higher resistance to in vitro degradation but normal affinity to KISS1R, thus suggestive of increased bioavailability as the mechanism by which these abnormal kisspeptins induce precocious puberty (90).
Recently in an Asian cohort, potentially regulatory polymorphisms, as rs5780218 and rs12998, in KiSS1 gene were significantly associated to genetic susceptibility to central precocious puberty in Chinese girls by single-locus analysis (91). Nevertheless, these findings are inconsistently reported in literature and require additional validation in functional studies.
A role for neurokinin B in the hypothalamic regulation was also demonstrated when genetic studies in patients from consanguineous families with hypogonadotropic hypogonadism were found to have missense mutations in TAC3 (encodes neurokinin B) and TACR3 (encodes neurokinin B receptor) (92). Other cases have been reported since (93-96).
There is also long-standing evidence for the role of opioid systems in reproduction. In 1980, Wilkes reported the localization of β endorphin in the human hypothalamus (97). Studies involving the administration of naloxone and naltrexone (opioid antagonists) to humans showed stimulatory effects on LH secretion (98,99), and other studies supported the notion that endogenous opioids play a role in the control of HPG axis (100-104). In 2007, it was demonstrated that dynorphin and kisspeptin are co-localized along with neurokinin B in the same hypothalamic neuronal population in sheep, therefore termed KNDy (Kisspeptin-Neurokinin B-Dynorphin) neurons, highlighting the possible interconnection between these neuropeptides in the control of GnRH and gonadotropin secretion (105-107). The co-localization of kisspeptin, neurokinin B and dynorphin has also been demonstrated in humans (108).
Kisspeptin neurons have also other important neuroanatomical relationships, such as with neuronal nitric oxide synthase neurons as demonstrated in prepubertal female sheep (109), or with somatostatin neurons in the rat hypothalamus (110).
Neuroanatomy of KNDy Neuronal Network
The location of kisspeptin neurons is different between rodents and human species. In humans, kisspeptin neurons are distributed in the rostral Pre-optic Area (POA) and in the infundibular nucleus in the hypothalamus () (108,111). In both male and female autopsy samples, the majority of kisspeptin cell bodies are identified in the infundibular nucleus, and a second dense population of kisspeptin neurons in the rostral POA (108). The infundibular nucleus (arcuate nucleus in non-human species) is similar across species, but the rostral region is more species specific (108,112,113). In rodents, the rostral population is located in the anteroventral periventricular nucleus (AVPV) and the periventricular nucleus (PeN), the continuum of this region named as the rostral periventricular region of the third ventricle (RP3V) (112,114). Humans and ruminants lack this well-defined RP3V population of kisspeptin neurons, which are more scattered within the preoptic region (113,115).
Kisspeptin axons form dense plexuses in the human infundibular stalk, where the secretion of GnRH occurs (108). Axo-somatic, axo-dendritic, and axo-axonal contacts between kisspeptin and GnRH axons were demonstrated at this level, showing that kisspeptin and GnRH networks are in close proximity (108,116). Moreover, GnRH neurons express Kiss1r mRNA, reinforcing the notion of kisspeptin involvement in GnRH secretion (117-119).
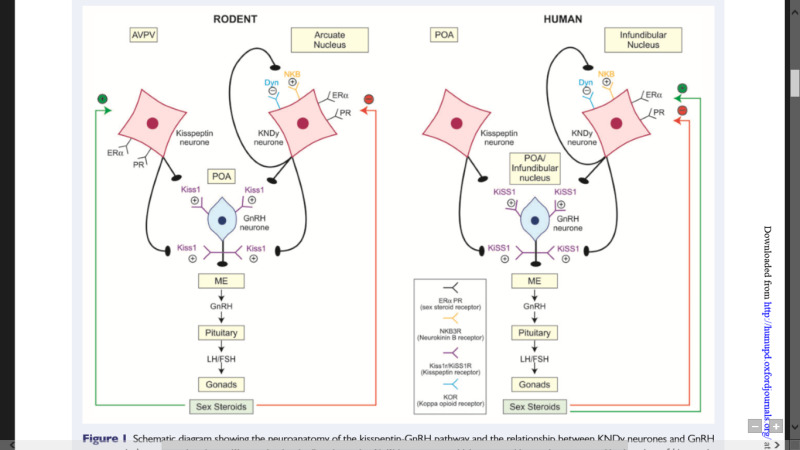
Figure 2.
Neuroanatomy of kisspeptin-GnRH pathway and the control of HPG axis in humans and rodents. Kisspeptin signals directly to GnRH neurons, which express KISS1R. The location of kisspeptin neurons within the hypothalamus is species specific, residing within the anteroventral periventricular nucleus (AVPV) and the arcuate nucleus in rodents, and within the preoptic area (POA) and the infundibular nucleus in humans. Kisspeptin neurons in the infundibular nucleus (humans)/arcuate nucleus (rodents) co-express neurokinin B and dynorphin (KNDy neurons), which autosynaptically regulate kisspeptin secretion (via neurokinin B receptor and kappa opioid peptide receptor). In humans, infundibular KNDy neurons relay negative (red) and positive (green) feedback, whereas in rodents the negative and positive steroid feedback are mediated via arcuate nucleus and AVPV respectively. The role of human POA kisspeptin neurons in sex steroid feedback is not yet clear. (Adapted from: Skorupskaite K, George JT, Anderson RA. The kisspeptin-GnRH pathway in human reproductive health and disease. Human Reproduction Update. 2014;20:485-500).
Three-quarters of kisspeptin-immunoreactive cells in the human infundibular nucleus of the hypothalamus co-express neurokinin B and dynorphin (KNDy neurons) (108,120). KNDy neurons in rodents and ruminants are localized in the arcuate nucleus of the hypothalamus. However, neurokinin B and dynorphin are absent from kisspeptin neurons in the hypothalamic POA () (67,115). This differential expression of neuropeptides may reflect distinct functions of these two kisspeptin populations with kisspeptin neurons in the AVPV acting as LH surge generators, while those in the ARC (including KNDy neurons) acting as LH pulse generators.
Significant kisspeptin expression was also demonstrated in central extra-hypothalamic sites, including in limbic and paralimbic brain regions, such as medial amygdala, cingulate, globus pallidus, hippocampus, putamen and thalamus, key areas of neurobiological control of sexual and emotional behaviors (reviewed in detail in (121)), as well as peripherally in organs like ovary, testis, uterus and placenta where the kisspeptin system may also play a part in reproduction function (122,123).
Apart from reproductive and central kisspeptin expression, the kisspeptin signaling system has been demonstrated in several peripheral tissues, namely, in pancreas (involved in glucose-stimulated insulin secretion); in endothelial cells of different vascular beds as coronary artery, aorta and umbilical vein (triggering vasoconstriction); in the kidney, namely, in tubular cells, collecting duct cells and vascular smooth muscle cells (involved in function and renal morphogenesis); as well as in bone, fat and liver tissue (78,124,125).
Interactions Between Kisspeptin, Neurokinin B and Dynorphin
KDNy neurons act synergistically to induce coordinated and pulsatile GnRH secretion by regulating the neuroactivity of other KDNy cells. This is supported by the existence of neurokinin B and kappa opioid peptide receptors (receptor for dynorphin) within the KNDy cells, but not kisspeptin receptors, which are predominantly expressed on GnRH neurons (107,120,126). Neuron-neuron and neuron-glia communications via gap junctions contribute for the synchronized activities among KNDy neurons (127).
Neurokinins (A and B) are members of the tachykinin family of peptides, which stimulate three related GPCRs (encoded by TACR1, TACR2 and TACR3) (128) This family also contains substance P, neuropeptide K, neuropeptide γ, hemokinin-1, and more recently endokinins. Neurokinin B acts predominantly on TACR3. Neurokinin B increases the membrane potential of KNDy neurons, leading to an increase in KNDy neuron pulsatile activity which, in turn, will promote the secretion of kisspeptin leading, ultimately, to GnRH secretion (67,129,130). Neurokinin B signaling regulates GnRH/LH secretion in healthy women, and it is crucial for the mediation of the estrogenic positive and negative feedback on LH secretion (131-133). There is rapidly increasing interest in the therapeutic value of neurokinin antagonists in several indications in reproductive health, recently reviewed in (134).
In women with polycystic ovary syndrome, the relationship between kisspeptin and gonadotropin levels has been widely explored in those with anovulatory cycles (135-138). Most studies have shown higher serum levels of kisspeptin and LH when the oligomenorrhea phenotype is present, despite the high heterogeneity observed. In this context, potential treatments targeting neuroendocrine dysfunction emerged. The administration of neurokinin 3 receptor antagonists markedly reduced serum LH concentration and pulse frequency, as well as serum testosterone (139-141). A recent study confirmed a complex crosstalk between neurokinin B and kisspeptin pathways in the regulation of GnRH secretion in polycystic ovary syndrome. In this study, kisspeptin-10 infusion given to women with polycystic ovary syndrome increased LH secretion with a direct relationship to estradiol exposure. Neurokinin 3 receptor antagonism reduced LH secretion and pulsatility, and whilst LH response to kisspeptin-10 was preserved, its relationship with circulating estradiol was not. More interestingly, although kisspeptin-10 increased LH pulse frequency, changes in other parameters of LH secretory pattern were prevented when co-administered with neurokinin 3 receptor antagonists (141).
In postmenopausal women, seven day treatment with neurokinin 3 receptor antagonist decreased LH secretion, but not FSH secretion, as well as lead to a remarkable reduction in hot flushes (142). Neurokinin 3 receptor antagonism efficiency in treating menopausal hot flushes has been also demonstrated in other clinical trials (143,144). Fezolinetant administrated in a single daily dose regimen (30 or 45mg/daily) for treatment of moderate to severe vasomotor symptoms reduced these symptoms by over 50% from baseline within the first week and persistently during the 52-week treatment period, and is now approved for the treatment of menopausal vasomotor symptoms (145-148).
A second comparable drug, elinzanetant, which differs in its pharmacology in that it is an antagonist at the NK1 as well as NK3 receptor, has also been recently demonstrated to reduce the severity and frequency of moderate-to-severe vasomotor symptoms and also to improve sleep quality and menopause-related quality of life (149). The importance of NK1 receptor antagonism in these effects is unclear.
In healthy men, neurokinin B signaling display a central role for the reproductive function, and this is functionally upstream of kisspeptin-mediated GnRH secretion: LH, FSH and testosterone secretion decreased during the administration of a neurokinin 3 receptor antagonist, while kisspeptin-10 administration restored LH secretion to the same degree before and during neurokinin 3 receptor antagonist treatment (150).
An increase in the expression of Kiss1 in the hypothalamic neurons was observed following senktide (agonist of neurokinin B) administration (151), and its stimulatory effects were abolished in Gpr54 knock-out male (152). In ovariectomized goats, neurokinin B stimulated LH secretion through electrical multi-unit activity corresponded to LH secretion, suggesting a hypothalamic site for this GnRH pulse generation (153). GnRH antagonists abolished the stimulatory effect of neurokinin B, demonstrating its site of action to be functionally higher than the GnRH receptor (154,155).
Dynorphins act as the decelerator that inhibits KNDy neurons pulsatility. Studies involving the administration of opioid antagonists to humans have shown stimulatory effects on LH secretion in late follicular and mid-luteal phase (98,99), and together with other studies (100-104), highlight the inhibitory input by dynorphins on kisspeptin signalling, and consequently on GnRH/gonadotropin secretion. Through the stimulatory effects of neurokinin B and kisspeptin, and the inhibitory action of dynorphin, these neuropeptides coordinate pulsatile GnRH and LH secretion () (156,157).
Kisspeptin-mediated GnRH secretion is sex steroid dependent. Estrogen and progesterone directly modulate kisspeptin activity though the sex-steroid receptors expressed on kisspeptin neurons at both AVPV and the arcuate nucleus (158-160). Furthermore, two distinct populations of kisspeptin neurons, the infundibular/arcuate region of which interacts with neurokinin B and dynorphin, appear to mediate distinct sex-steroid pathways (discussed in more detail in sections 4.1-4.4).
Briefly, in humans, KNDy neurons in the infundibular nucleus alone are involved in negative and positive sex-steroid feedback, whereas in rodents positive sex-steroid feedback seems to be mediated via kisspeptin neurons in the AVPV region and negative sex-steroid feedback via the arcuate KNDy neurons () (67,111,160,161).
Stimulatory Effect of Kisspeptin on GnRH and Gonadotropin Secretion
Kisspeptin is a potent stimulator of the HPG axis – and in fact, it is the most potent GnRH secretagogue currently known. Kisspeptin signals directly to the hypothalamic GnRH neurons via kisspeptin receptor to release GnRH into the portal circulation, which in turn stimulates the anterior pituitary gonadotropes to produce LH and FSH (129,162).
The stimulatory effects of kisspeptin on LH secretion have been documented in animal models (163-166). This is consistent with human studies, where kisspeptin increases both LH and FSH secretion with the preferential stimulatory effect on the former (67,167-177). Kissppetin-54 was first administered in healthy men as an intravenous infusion with dose-dependent rise in LH secretion (169). Since then kisspeptin was administered in different isoforms (kisspeptin-54 and kisspeptin-10), different routes (subcutaneous and intravenous), different types of exposure (continuous and bolus), to healthy men and women and in endocrine disease models with low gonadotropin output, all showing stimulatory effect of kisspeptin on LH secretion (fully reviewed in (67)).
Pulsatile GnRH secretion correlates with LH pulsatility, prompting investigation of the effect of kisspeptin on regulating LH pulse frequency. LH pulse frequency and amplitude were increased following intravenous infusion of kisspeptin-10 in healthy men (172), and subcutaneous bolus of kisspeptin-54 in healthy women (174). The hypothalamic response to kisspeptin-54 and the pituitary response to GnRH are preserved in healthy older men (178). Kisspeptin also stimulates LH pulse frequency in reproductive endocrine disorders of low LH pulsatility, including hypothalamic amenorrhea, defects in the neurokinin B pathway and hypogonadal men with diabetes (96,179,180). Indeed, kisspeptin-54 and kisspeptin-10, as well as kisspeptin agonists like MVT-602 (previously known as TAK-448) are able to stimulate physiological reproductive hormone secretion in individuals with functional hypogonadism related to deficient GnRH secretion, such as in hypothalamic amenorrhea or polycystic ovary syndrome (181,182).
In addition, recent findings have explored further the effects of MVT-602. LH concentration increased in a dose-dependent manner, resembling the amplitude and duration found in the physiological mid-cycle LH surge, proving to be safe and well tolerated throughout the dose range (0.3 – 3.0 mg) (183). This approach to mimicking the physiological response during oocyte maturation and ovulation may have clinical utility for women during medically assisted reproduction.
Kisspeptin regulates GnRH and subsequently gonadotropin secretion through Kiss1r, as suggested by Messager who demonstrated no detectable LH levels in response to kisspeptin in Kiss1r knockout mice (119). The prevention of the stimulatory effect of kisspeptin on LH secretion by GnRH antagonists indicate that kisspeptin action is GnRH-mediated (118,164,184-186). This is further supported by the observation that kisspeptin cause depolarization of GnRH neurons (117) and stimulate GnRH release from hypothalamic explants (187,188). The expression of GnRH mRNA is upregulated in GnRH neurons following kisspeptin administration (189). Moreover, in patients with impaired functional capacity of GnRH neurons (idiopathic hypogonadotropic hypogonadism), the same dose of kisspeptin failed to induce LH response seen in healthy men and women (190). In female rats, ablation of KNDy neurons resulted in hypogonadotropic hypogonadism, confirming its role in the maintenance of normal LH levels and to estrous cyclicity (191).
Some investigators have demonstrated a direct stimulatory effect of kisspeptin on gonadotropes, but this direct stimulatory action of kisspeptin on gonadotropes remains debatable (192-196). Kiss1 and Kiss1r gene expression has been shown in gonadotropes, and gonadotropin secretion from the pituitary explants was observed following exposure to kisspeptin (78,192-195). Moreover, LHβ and FSHβ gene expression was upregulated in the primary pituitary cells treated with kisspeptin. Whilst kisspeptin can directly regulate gonadotropins at the transcriptional level, it appears to be less relevant than the GnRH-mediated action (67,195,196).
Desensitization Effect of Chronic or Continuous Exposure to Kisspeptin
Continuous administration of GnRH desensitizes the HPG axis by downregulation of GnRH receptors and desensitization of gonadotropes, following an initial stimulatory effect (39). It is therefore important to ascertain the effects of continuous exposure to kisspeptin on the HPG axis. Efforts have been made to assess the impact of continuous infusions of kisspeptin in a number of animal experiments (119,197-200).
In adult rats, continuous administration of kisspeptin-54 increased serum LH and free testosterone on day one, but this stimulatory effect was lost after 2 days, indicative of kisspeptin receptor desensitization (200). In rhesus monkeys, the continuous administration of kisspeptin-10 resulted in suppression of LH secretion, indicating desensitization of kisspeptin receptor (198). The kisspeptin receptor has been shown to desensitize in vitro (197). In sheep, infusion of kisspeptin-10 resulted in acute increase in serum LH levels, which declined by the end of 4-hour infusion, while GnRH remained elevated following the discontinuation of kisspeptin-10 administration. This suggests that desensitization to GnRH could be occurring at the level of pituitary gonadotropes (119).
Consistent with animal studies, Jayasena et al. demonstrated that in women with hypothalamic amenorrhea an initial increase in LH and FSH secretion was not sustained following twice daily subcutaneous kisspeptin-54 administration for two weeks (176). Other studies in humans employing continuous or repeated kisspeptin administration provide conflicting evidence for kisspeptin-mediated desensitization and appear to be dose-related (172,180). High doses of kisspeptin may induce desensitization, but this is not apparent at lower doses (67). Sustained LH secretion and increased LH pulsatility was demonstrated with lower dose of kisspeptin-54 (0.01-1nmol/kg/h) infusion for 8 hours in women with hypothalamic amenorrhea (180) and kisspeptin-10 (3.1 nmol/kg/h) infusion for 22.5 hours in healthy men (172). In contrast, LH secretion was not maintained in three healthy men during the 24 hour infusion of kisspeptin-10 at 9.2 nmol/kg/h (the highest dose used in humans so far), although serum LH did not fall to the castrate levels and remained well above baseline at end of infusion (201).
Kisspeptin receptor agonist analogues, TAK-488 and TAK-683, induce desensitization when administered to healthy men (202,203). However, the ability of natural kisspeptin fragments to downregulate the HPG axis in humans remains to be established, and is to date complicated by differences in study protocols, in terms of isoform of kisspeptin used, duration (8 hours-2 weeks), mode and route of kisspeptin administration, lower doses of kisspeptin in human studies compared to animal, and the endocrine profile of the study participants (men versus women versus hypothalamic amenorrhea).
Sexual Dimorphism in Kisspeptin Signaling
The response to kisspeptin is different in men and women. In men, kisspeptin potently stimulates the release of LH, but in women the effect of kisspeptin is variable and dependent on the phase of menstrual cycle (67). Whilst men respond to the modest doses of kisspeptin, LH response to kisspeptin in healthy women is minimal and inconsistent in the early follicular phase but greatest in the pre-ovulatory phase of the menstrual cycle (169-171,177). This indicates that in addition to the fluctuations in sex-steroid milieu, other mechanisms, such as changes in pituitary sensitivity to GnRH or GnRH network responsiveness to kisspeptin regulate the sensitivity to kisspeptin throughout the menstrual cycle (67,126,204).
Not only there is sexual dimorphism in gonadotropin response to kisspeptin, but there are also anatomical differences. Female hypothalami have significantly more kisspeptin fibers and kisspeptin cell bodies than men (173). Only a few kisspeptin cell bodies are present in the male infundibular nucleus and none in the rostral periventricular nucleus, which is on contrary to the female hypothalami with abundant kisspeptin network in both of these hypothalamic nuclei (108). These sex differences in kisspeptin neurons appear to be established early during perinatal development through the action of sex steroids (126,205).
These marked functional and anatomical differences may reflect sexually dimorphic roles of kisspeptin between both sexes, influencing their reproductive functions, namely the sex steroid feedback in GnRH and gonadotropin secretion (67).
Kisspeptin, GnRH and Puberty
Kisspeptin is crucial for normal pubertal development, the discovery of which formed the basis for the obligate role of kisspeptin signaling in the control of reproductive function (206). More than a decade ago two independent groups identified ‘inactivating’ mutations in KISS1R in patients with hypogonadotropic hypogonadism presenting with pubertal delay (85,86). Recently, a male patient with a biallelic loss-of-function KISS1R mutation was described who had undergone a normal and timely puberty, although as a child he had presented with microphallus and bilateral cryptorchidism. This suggests different levels of dependence of the hypothalamic-pituitary-gonadal axis on kisspeptin signaling during the reproductive life span, with the mini-puberty of infancy appearing more dependent on the kisspeptin system than is adolescent puberty (207). On the other hand, activating mutations in KISS1R and KISS1 were then described in children with central precocious puberty (89,90).
Hypothalamic expression of Kiss1 and Kiss1R mRNA is upregulated at puberty (117,165,208), and the percentage of GnRH neurons depolarizing in response to kisspeptin increases from juvenile (25%) to pubertal (50%) and to adult mice (>90%) (117), suggesting that GnRH neurons may acquire sensitivity to kisspeptin across puberty. In monkeys, kisspeptin-54 secretion and pulsatility increased at the onset of puberty (209). Moreover, the exogenous administration of kisspeptin resulted in earlier puberty in rats and monkeys (208,210), whereas kisspeptin antagonists delayed puberty in rats (186) and inhibited GnRH release in pubertal monkeys (211). In other study, daily injections of a synthetic kisspeptin analogue have been shown to significantly advance puberty in prepubertal female mice (212). GnRH neuron-specific Kiss1r knockout mouse showed a delay in pubertal onset, abnormal estrous cyclicity in female and abnormal external genitalia in male (microphallus, decreased anogenital distance associated with failure of preputial gland separation) (213).
Exogenous kisspeptin stimulated GnRH-induced LH secretion in patients with hypogonadotropism resulted in a spontaneous and permanent activation of their hypothalamic-pituitary-gonadal axis, whereas patients with idiopathic hypogonadotropic hypogonadism and no spontaneous LH pulsatility did not respond to kisspeptin, suggesting that the reversal of hypogonadism, sexual maturation and puberty may well be associated with the acquisition of kisspeptin responsiveness which in turn signals the emergence of reproductive endocrine activity (214). A recent study, 15 children with delayed puberty were administered intravenous kisspeptin and displayed divergent responses, with seven subjects having no response to kisspeptin, whereas others having either robust response (comparable to those of adults) or intermediate responses as perceived in one case (215).
GnRH release during puberty appears to require a cooperative mechanism between the kisspeptin/NKB networks in close interaction with different neuropeptides, as substance P, NKA, RFRP-3 and alpha-MSH, working as partners to regulate puberty timing influenced, naturally, by a combination of genetic, environmental, and gene-environment interactions (216).
Agonists and antagonists of kisspeptin and NKB were administered into the stalk-median eminence (region with high concentration of GnRH, kisspeptin and NKB neuroterminal fibers), and it was found that both kisspeptin-10 and the NK3R agonist senktide stimulated GnRH release in a dose-responsive manner in prepubertal and pubertal monkeys. However, senktide-induced GnRH release was blocked in the presence of a KISS1R antagonist and the kisspeptin-induced GnRH release was blocked in the presence of NK3R antagonist in pubertal monkeys, leading to the notion that a reciprocal signaling mechanism between kisspeptin and NKB exists and is possibly necessary for a normal puberty (217). These data together emphasizes that disrupted kisspeptin-GPR54-NKB signaling leads to hypogonadotropic hypogonadism, reinforcing the critical role of kisspeptin in puberty.
REGULATION OF GnRH AND GONADOTROPIN SECRETION
Development and maintenance of normal reproductive function requires a coordinated interplay between neuroendocrine, metabolic, and environmental factors. The GnRH-gonadotropin system plays a central role in the regulation of reproduction by integrating different signals and factors () (126,204).
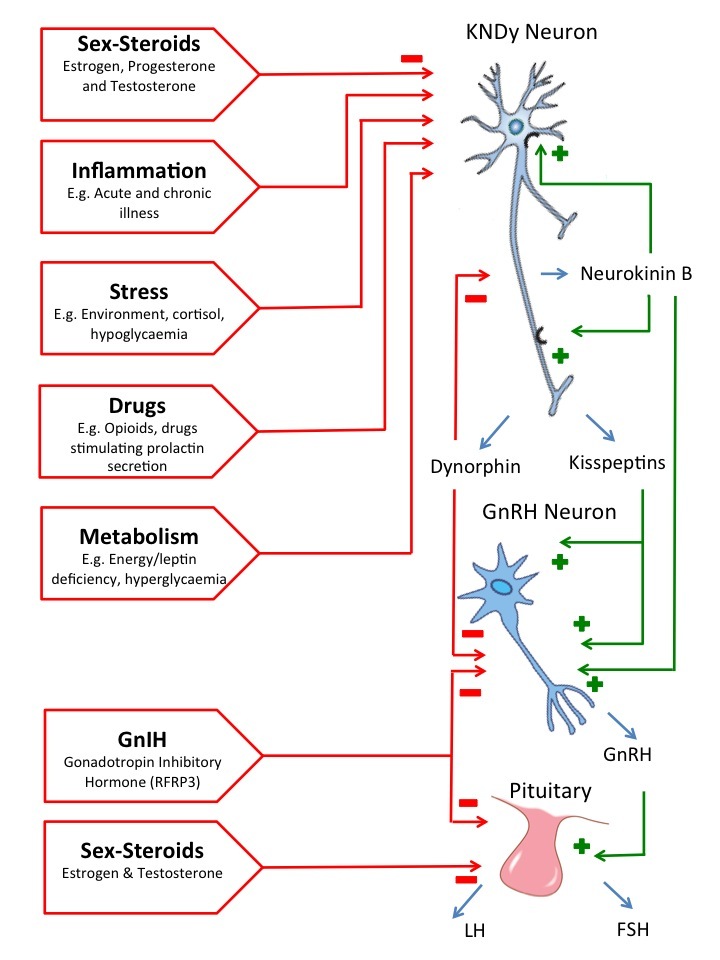
Figure 3.
Neuroendocrine regulation of GnRH/gonadotropin secretion. The GnRH-gonadotropin system plays a central role in the regulation of reproduction by integrating different neuroendocrine, metabolic and environmental signals/factors. The KNDy signaling has a key role in this process by integrating some of these signals and by regulating GnRH neurons.
Overview of Sex Steroid Feedback
A crucial role for sex steroids in the regulation of GnRH neurons and/or gonadotropes in humans was initially proposed as serial blood sampling and gonadotropin assays in women through phases of menstrual cycle showed an uneven distribution, with a clear mid-cycle surge in LH and FSH. Two mechanisms were proposed to mediate this effect: first, GnRH secretion is altered in response to the steroid milieu; second, sensitivity of the gonadotropes to a GnRH input is sex-steroid dependent, although the exact mechanism remains controversial due to inter-species variation (218).
Hypothalamic secretion of GnRH increases during proesterus in rats (219), sheep (220), and non-human primates (221). Pulsatile once hourly administration of exogenous GnRH restored ovulation in Rhesus monkeys with hypothalamic lesions which abolished GnRH secretion, suggesting that it was the ‘ebb and flow’ of ovarian estrogen feedback acting directly on the pituitary which triggered an LH surge (222). In humans, endogenous GnRH secretion is potentially diminished during the pre-ovulatory LH surge and the suppression of gonadotropin secretion is greater with lower doses of a GnRH receptor antagonist during the mid-cycle surge in comparison to the other phases of the menstrual cycle (223). This suggests that pituitary gonadotrope sensitivity to GnRH is enhanced during the mid-cycle surge. Administration of exogenous estradiol or testosterone in men with hypogonadotropic hypogonadism receiving pulsatile GnRH therapy, decreased gonadotropin concentrations, demonstrating inhibitory effects of sex-steroids at the level of pituitary (224). A direct effect of estrogen on gonadotropes is further demonstrated by the inhibition of LH secretion from rat pituitary gonadotropes in vitro (225). Literature to date suggests that there is dual-site sex-steroid feedback in the regulation of gonadotropin secretion, occurring at the level of both pituitary and hypothalamus (226-231).
Estrogen Feedback
Patterns of GnRH and LH secretion across the menstrual cycle are modulated by estradiol feedback. A biphasic effect of estradiol on gonadotropin secretion has long been established and it is essential for normal menstrual cycle, with an initial negative feedback (greater suppression of FSH) and a subsequent positive feedback (more prominent for LH) (32). However, the basis for estrogen feedback has been unclear for a long time. GnRH neurons do not express estrogen receptor alpha (ER-alpha) (232,233), and therefore a mediator between gonads and hypothalamus was missed. Recent evidence suggests that kisspeptin and neurokinin B (132) appears to be providing this “missing link” as a key regulator of both negative and positive estrogen feedback (67,126).
KNDy neurons in the infundibular nucleus in humans and the arcuate nucleus in other mammals mediate negative estrogen feedback. Estrogen suppresses kisspeptin and neurokinin B release from KNDy neurons, which reduce their stimulatory input to GnRH neurons. Simultaneously, there is a relative deficiency in dynorphin signaling as part of this negative feedback, releasing the inhibitory action on kisspeptin signaling () (67). Immunohistochemical staining of the postmenopausal women hypothalami showed up-regulated expression of KISS1 mRNA and hypertrophy of kisspeptin neurons in the infundibular nucleus when compared to the premenopausal women (111). These hypertrophied kisspeptin neurons co-localized with ER-alpha, had increased expression of neurokinin B and decreased levels of prodynorphin mRNA (234-236). The above evidence for the involvement of the infundibular KNDy system in mediating negative estrogen feedback in humans is consistent with animal studies. Kisspeptin neurons in the arcuate nucleus show frequent co-localization with ER-alpha (160,237). In ovariectomized animals, the expression of Kiss1 and neurokinin B mRNA was up-regulated but prodynorphin mRNA reduced in the arcuate nucleus (equivalent to the infundibular nucleus in humans), and this was reversed by estrogen replacement (102,115,120,238-242). Postmenopausal women are resistant to the stimulatory effect of kisspeptin on LH secretion (142,243), but postmenopausal women receiving estradiol replacement therapy are only resistant to kisspeptin initially and then they do demonstrate a remarkable increase in LH pulse amplitude with direct correlation to the circulating levels of estradiol and duration of kisspeptin administration (243). However, neurokinin B regulates gonadotropin secretion in postmenopausal women, and antagonizing the neurokinin 3 receptor modestly decreases LH secretion in this context (142).
Interestingly, the use of neurokinin 3 receptor antagonists has been shown to effectively reduce the frequency and severity of menopause-related vasomotor symptoms owing to their inhibitory effect in the hypothalamic thermoregulatory center, and thus presenting a potential non-hormonal treatment option for menopausal women (144,148,149).
Negative estrogen feedback switches to positive feedback in the late follicular phase of menstrual cycle, in order to induce the pre-ovulatory LH surge. Ovarian estradiol seems to be the predominant signal to trigger this switch, via ER-alpha, stimulating RP3V kisspeptin neurons while it inhibits arcuate kisspeptin neurons. Recent evidence supports the role of kisspeptin in generating the LH surge: during an assisted conception cycle, kisspeptin-54, used instead of a routinely administered human chorionic gonadotropin, induced an LH surge, and oocyte maturation, with a subsequent live term birth (241). Repeated twice-daily administration of kisspeptin-54 shortened the menstrual cycle, suggesting that the onset of LH surge was advanced (173). This is further supported by antagonistic studies in animal models, where the administration of kisspeptin antiserum or antagonists blunt/prevent LH peak, whilst kisspeptin advances LH surge (211,244,245).
However, kisspeptin-mediated positive estrogen feedback has marked anatomical variations between humans and other species. In rodents, positive estrogen feedback is mediated via the AVPV nucleus, which is absent in humans, other primates and sheep (). AVPV neurons are sexually dimorphic, with higher density of ER-alpha described in females and AVPV kisspeptin neurons, as a subset of AVPV neurons, share this pattern (246). There seems to be functional specialization, since only a subset of AVPV kisspeptin neurons (~1/3) are synaptically connected to GnRH cell bodies, but of these, nearly all express estrogen sensitivity and most co-express tyrosine hydroxylase to facilitate positive feedback (247). The expression of Kiss1 mRNA in the AVPV nucleus is low following an ovariectomy but is dramatically increased with estrogen treatment and at the time of LH surge (160,161). In sheep, positive estrogen feedback is mediated though the arcuate nucleus, where the expression of Kiss1 mRNA is the greatest at the pre-ovulatory LH surge (195).
There are no studies looking at the anatomical region of estrogen mediating positive feedback in humans. Although there does not appear to be two distinct anatomical populations of kisspeptin neurons to relay negative and positive sex-steroid feedback in humans, it is possible that separate signaling pathways exists to mediate gonadal steroid feedback.
Whilst it is clear that kisspeptin is involved in estrogen-induced mid-cycle gonadotropin surge, the role of KNDy neurons in positive estrogen feedback is less obvious. In sheep, the expression of neurokinin B mRNA was increased during the LH surge, and neurokinin B receptor agonist senktide induced LH secretion mimicking its mid-cycle surge (248,249). However, this has not been reproduced in other species, including humans (180). In summary, KNDy neurons mediate negative estrogen feedback in the infundibular nucleus in humans and the arcuate nucleus in other species. Positive estrogen feedback is mediated via kisspeptin neurons, which show marked inter-species anatomical variation.
In addition to the gonads, the brain is one of the major organs producing estradiol, and recently a number of studies demonstrated that estradiol is synthesized and released in the hypothalamus (i.e. neuroestradiol) contributing to the regulation of GnRH release, particularly regarding its positive feedback effect during the preovulatory GnRH/LH surge (250).
Progesterone Feedback
Progesterone reduces LH pulse frequency in healthy women. LH secretory pattern in women exposed to exogenous progesterone was comparable to LH profile observed in the mid-luteal phase, demonstrating that progesterone plays a central role in the luteal phase of menstrual cycle (251). These inhibitory effects of progesterone on gonadotropin secretion are mediated by the progesterone receptor (PR) (252). The suppressive effect of progesterone on LH secretion was diminished in the context of estrogen deficiency, while co-administration of estradiol restored it (252), suggesting an interplay between these sex steroids. However, the presence of PR on only a small subset of GnRH neurons (253-255) led to the notion that intermediaries are involved in mediating inhibitory progesterone signal to GnRH neurons.
There is evidence that KNDy neurons play a role in mediating progesterone feedback on GnRH through dynorphin signaling () (102,120). PR have been demonstrated to be co-localized with dynorphin in the KNDy neurons (159) and progesterone increased dynorphin concentrations (256). Moreover, the number of preprodynorphin mRNA expressing cells decreased in postmenopausal women (236) and in ovariectomized ewes, but normalized with exogenous progesterone to luteal levels (256).
Testosterone Feedback
Testosterone exerts negative feedback on gonadotropin secretion. Early studies verified that LH and FSH pulse frequency are enhanced in hypogonadal men and exogenous testosterone decreases gonadotropin secretion, suggesting that testosterone have an inhibitory effect on GnRH secretion (230,257).
Few GnRH neurons express androgen receptors (AR) (258). GnRH neurons were thus considered to be reliant on an intermediary neuronal population to mediate testosterone feedback. A key role for KNDy neurons in this mediation has been suggested, as these neurons express AR which directly mediate the androgen feedback. The androgen feedback may also rely on the aromatization of testosterone, as testosterone-induced suppression of Kiss1 mRNA in the rodent arcuate nucleus is identical to that observed with estradiol, but more than that observed with dihydrotestosterone administration (259). The cross-talk between AR and ER was suggested from animal studies: AR expression was downregulated in the prostate following neonatal estrogen exposure (260), and AR transcription was modulated following a co-transfection of AR and ER (261).
Navarro has described a role for KNDy neurons in mediating the negative testosterone feedback on GnRH secretion, and provided evidence that neurokinin B released from KNDy neurons is part of an auto-feedback loop that generates the pulsatile secretion of Kiss1 and GnRH in male mice: Kiss1 and dynorphin mRNA are regulated by testosterone through estrogen and androgen receptor-dependent pathways; KNDy neurons express neurokinin B receptor whereas GnRH neurons do not, and senktide (an agonist for the neurokinin B receptor) activates KNDy neurons leading to gonadotropin secretion but has no discernible effect on GnRH neurons (262). Other studies demonstrated that the suppression of gonadotropin secretion using testosterone is associated with a reduction of Kiss1 mRNA in the hypothalamus (118,208,263). Moreover, post-orchidectomy rise in LH in rodents can be blocked by kisspeptin antagonists, further suggesting that kisspeptin system mediates the hypothalamic androgen feedback (186).
Stress and Glucocorticoids
Physical and psychological stress is associated with hypothalamic amenorrhea, possibly though the activation of hypothalamic-pituitary-adrenal (HPA) axis (264,265). Experimental evidence points towards a cortisol-mediated suppression of gonadotropin secretion as the main key pathway to explain stress-induced gonadotropin suppression (55,266-273). The negative effect of cortisol on HPG axis is recognized to occur at both pituitary and hypothalamic levels. There are also data suggesting that upstream factors in the HPA axis, such as Corticotropin Releasing Hormone (CRH) and vasopressin may play a mediatory role (274,275).
Cortisol secretion in women with hypothalamic amenorrhea is elevated (267), and evening adrenocorticotropic hormone (ACTH) and cortisol concentrations are higher in excessive exercise (266,270). Administration of exogenous glucocorticoids to eugonadal women was associated with a decrease in LH pulse frequency, suggesting that glucocorticoids have a negative action on GnRH secretion (273). In ovine portal blood, cortisol administration led to a decrease in GnRH pulse frequency (272). Inferences of cortisol effects on gonadotropin secretion were also derived from observations in women and men with Cushing’s syndrome (condition associated with excessive cortisol secretion), where exogenous GnRH preferentially stimulates FSH whilst LH remains unchanged (268,271). The resolution of male hypogonadotropic hypogonadism was also observed in men with remission of Cushing’s disease (271). This negative input of cortisol on the HPG axis may be modulated by sex-steroid hormones, and kisspeptin signaling has also been implicated in the process.
Cortisol alone had no impact on GnRH pulsatility in ovariectomized ewes, but the co-administration of estradiol and progesterone led to a 70% decrease in GnRH secretion (272). Decreased hypothalamic Kiss1 mRNA expression has been observed during exposure to stress or exogenous glucocorticoids. The role of kisspeptin in mediating stress inputs is further supported by the expression of glucocorticoid receptor on murine kisspeptin neurons (276). Colocalization by immunohistochemistry of CRH receptor (CRH-R) in most hypothalamic kisspeptin neurons in the AVPV/PeN and ARC nuclei as well as glucocorticoid receptor (GR) in AVPV/PeN kisspeptin neurons support a relevant direct role of kisspeptin neurons in the inhibitory effects of CRH/ glucocorticoids (277).
Hypothalamic CRH neurons, important regulators of the stress response, also directly modulate GnRH excitability in a dose-dependent and receptor-specific manner, and the GnRH response to CRH is influenced by estrogens (278). Intracerebroventricular administration of CRH in female rats suppressed LH pulsatility and the LH surge, and this suppression was enhanced by estrogens (279).
Animal models have also linked increased exposure to RFamide-related peptide-3 (RFRP-3) during acute and chronic stress and hypothalamic expression of GnIH mRNA. Along these lines, the surface of GnIH neurons has glucocorticoid receptors and hydrocortisone administration was associated to an increased GnIH mRNA expression, ultimately leading to lower GnRH activity and dysregulation of the HPG axis (280). Together, these findings emphasize that kisspeptin as GnIH provide relevant inputs that contribute to an inhibitory effect of corticosteroids on gonadal axis during stress.
Prolactin
Prolactin is a well-known inhibitor of GnRH release and a suppressor of the HPG axis. The association between hyperprolactinemia and reproductive dysfunction has long been established, accounting for 14% of secondary amenorrhea and hypogonadism cases (281) and for a third of women presenting with infertility (282,283). Hyperprolactinemia is evident in 16% of men with erectile dysfunction and in 11% of men with oligospermia (284). The decreased pulsatility of LH in hyperprolactinemia responds to bromocriptine (285). GnRH therapy has restored ovulation and normal luteal function in bromocriptine resistant hyperprolactinemia women (286,287), suggesting that prolactin exerts inhibition through direct reduction of GnRH secretion.
The neuroendocrine pathway by which prolactin inhibits GnRH pulse frequency remains to be fully elucidated. A direct action of prolactin on the GnRH neuronal network is possible (288,289). Prolactin has also been demonstrated to influence other systems, including GABA (290), β endorphins (291), neuropeptide Y (292) and dopaminergic systems (via tuberoinfundibular dopamine (TIDA) neurons) (293).
Nevertheless, data suggest that prolactin receptors are expressed in most kisspeptin neurons but only in a small proportion of GnRH neurons, indicating that kisspeptin signaling may have a role in this context (288,294). In rodent models, kisspeptin neurons in the arcuate nucleus modulate dopamine release from dopaminergic neurons, thereby regulating prolactin secretion (295). Kiss1 expression is decreased in lactation, a physiological state associated with hyperprolactinemia (296). Prolactin-sensitive GABA and kisspeptin neurons were identified in regions of the rat hypothalamus (294). Moreover, in a mouse model of anovulatory hyperprolactinemia (induced by a continuous infusion of prolactin), Kiss1 mRNA levels were diminished and peripheral administration of kisspeptin restored gonadotropin secretion and ovarian cyclicity (297). There are also other animal studies reporting an inhibitory effect of prolactin on Kiss1 expression (298,299). This data suggests that kisspeptin is a possible link between hyperprolactinemia and GnRH deficiency. The administration of kisspeptin-10 reactivated the gonadotropin secretion in women with hyperprolactinemia-induced hypogonadotropic amenorrhea, suggesting that GnRH deficiency in the context of hyperprolactinemia is, at least in part, mediated by an impaired hypothalamic kisspeptin secretion (300).
On the other hand, kisspeptins appears to have a stimulatory effect on prolactin release, as demonstrated in a recent study in ovariectomized rats which had intracerebroventricular injections of kisspeptin-10 with subsequent increase in prolactin release, and this required the estrogen receptor-alpha and was potentiated by progesterone via progesterone receptor activation (301).
Nutrition and Metabolism
A link between energy balance and reproductive function enables organisms to survive to reproductive maturity and to withstand the energy needs of parturition, lactation, and other parental behaviors. This link optimizes reproductive success under fluctuating metabolic conditions (302). Kisspeptin signaling may link nutrition/metabolic status and reproduction by sensing energy stores and translate this information into GnRH secretion (303). These relations elucidate further associations between reproductive dysfunction and metabolic disturbances, such as diabetes, obesity or anorexia nervosa (67,304,305).
Food deprivation impairs GnRH and gonadotropin secretion, and leptin (a satiety hormone secreted by adipose tissue, the levels of which drop in response to fasting) plays a role in this inter-regulation by stimulating LH release (67,306-308). Periods of fasting and calorie restriction decrease LH pulse frequency and increase pulse amplitude (302,309-311). Administration of recombinant leptin increased LH pulse frequency in women with hypothalamic amenorrhea (312) and prevented fasting-induced drop in testosterone and LH pulsatility in healthy men (313). Moreover, humans with mutations in leptin or in leptin receptor show hypogonadism (314). Thus, the crosstalk between kisspeptin and leptin is relevant for reproduction and fertility (71), including in the setting of assisted reproduction techniques (315).
Kisspeptin neurons may have a role in mediating the metabolic signals of leptin on the control of HPG axis, as 40% of the arcuate kisspeptin neurons express leptin receptors in contrast to the GnRH neurons, where leptin receptors are absent (316-319). Food deprivation is associated with a decrease in kisspeptin, and subsequent reduction in gonadotropin secretion (320-323). Levels of low Kiss1 mRNA expression in the leptin-deficient ob/ob mice are partially upregulated by exogenous leptin (161). Moreover, exogenous kisspeptin restored vaginal opening (marker of sexual maturation) in malnourished rodents (320). Animal models of type 1 diabetes, characterized by insulin deficiency and impaired cellular nutrition, had hypogonadotropic hypogonadism and decreased Kiss1 mRNA expression. Repeated administration of kisspeptin to these rodents increased prostate and testis weight (324). It is plausible that a relative deficiency of kisspeptin secretion is a mechanism for hypogonadotropic hypogonadism in patients with obesity and diabetes (179). In hypogonadal men with type 2 diabetes, kisspeptin-10 increased LH secretion and pulse frequency (179). Although early studies appeared to suggest a direct link between kisspeptin and leptin, it seems that the neuronal pathway whereby leptin modulates GnRH is far more complex (325,326). Only partial restoration in Kiss1 mRNA in leptin-deficiency and normal pubertal development and fertility observed in selective leptin receptor deletion from kisspeptin neurons suggest that kisspeptin may link reproduction and metabolism through other ways than leptin (161,327). Proopiomelanocortin (POMC), agouti-related peptide, neuropeptide Y, ghrelin, and cocaine- and amphetamine-regulated transcript (CART) expressing neurons have been linked to this process (303,319). Kisspeptin neurons communicate with POMC and neuropeptide Y neurons and are able to modulate the expression of relevant genes in these cells (316). This link between kisspeptin and other peptides classically associated to food intake (as POMC and neuropeptide Y) was explored due to the anorexigenic effect of intracerebroventricular administration of KP-10 in male rats mediated via anorectic neuropeptides, nesfatin-1 and oxytocin, expressed in various hypothalamic nuclei. Diminished food intake and anorexia was significantly abolished by pretreatment with oxytocin receptor antagonist (328,329).
Several studies have also suggested that ghrelin can interact directly with hypothalamic neurons leading to suppression of gonadotropins release, and thus impairing fertility, an effect that is dependent of the estradiol milieu (303,330-332).
GABA (Gamma-Amino Butyric Acid)
GABA has also been implicated as a regulator of GnRH secretion. Although GABA is classically an inhibitory neurotransmitter in the central nervous system, most mature GnRH neurons are stimulated by GABA, which has attributed to GABA an excitatory action in HPG axis. The precise physiology of this mechanism is still unclear (333-337), but it may be related to the bidirectional interactions between GABA and kisspeptin pathways, as well as between these and GnRH neurons, in a variety of ways throughout development (338). In early development, GABA seems to increase KISS1 expression in embryonic phase and early postnatally, while in the absence of GABAergic input the expression of KISS1 declines (338,339). In the prepubertal period, the central restraint on GnRH secretion seems to be mediated by GABA possibly acting directly via kisspeptin neurons (338). In the peri-pubertal phase, the antagonism of GABA and the intrinsic disinhibition of kisspeptin neurons seem to be critical in puberty initiation and development (340,341). In adulthood, the interactions between GnRH-GABA-kisspeptin become more complex with HPG axis function critically dependent on such interactions. For instance, the preovulatory surge does not occur in the absence of GABA signaling, thus neurons co-expressing GABA and kisspeptin seem crucial in providing double excitatory input to GnRH neurons at the time of ovulation (338,342).
Additionally, in healthy men, total endogenous GABA levels in the anterior cingulate cortex, a key limbic structure, significantly decreased after intravenous infusion of kisspeptin (1 nmol/kg/h) demonstrating a potent inhibitory effect of kisspeptin on GABA levels which could be a fundamental concept in understanding the central limbic effect of kisspeptin in the human brain (343).
Other Neuropeptides
In addition to KNDy system and GABA, other peptides and neurotransmitters have been shown to influence GnRH-gonadotrope system: vasoactive intestinal polypeptide (VIP), vasopressin, catecholamines, nitric oxide, neurotensin, gonadotropin-inhibitory hormone (GnIH) /RFamide related peptide-3 (RFRP-3) (337), nucleobindin-2/nesfatin-1 (344). Excitatory inputs to the HPG axis may be mediated by VIP, catecholamines, glutamate and possibly vasopressin, whereas GnIH in birds, or its mammalian homolog RFRP-3, provide inhibitory inputs (345-349). RFRP neuronal populations have been detected mainly in the hypothalamic dorsomedial nucleus or adjacent regions, and they have projections to several hypothalamic areas including the arcuate nucleus, paraventricular nucleus, ventromedial nucleus and the lateral hypothalamus, all areas with major roles in the regulation of reproduction and energy balance (350,351). RFRP-3, encoded by the gene Rfrp, inhibits the electric firing of GnRH and kisspeptin neurons (346,352), which results in a suppression of GnRH-induced gonadotropin release with consequent inhibition of the reproductive axis (353). This RFRP-3 inhibitory input on the gonadotropin release is influenced by estrogens and may well be involved in their negative feedback. Estrogens reduce RFRP-3 expression and RFRP-3 neuronal activation (354,355).
Particular attention has been paid to the role of glutamate as a stimulatory modulator of the activity of ARC kisspeptin neurons, reaffirming the role of kisspeptin as a major neural integrator of inputs to GnRH neurons. Data from Kiss1 KO rats showed failure to increase GnRH/LH secretion following monosodium glutamate/NMDA administration (356).
link





